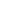Hse06 Vasp Online
KPOINTS: Automatic generation 0 Gamma 4 4 2 0 0 0
If your system has less than 50 atoms and you care about the band gap to 0.1 eV accuracy, pay the cost. If you're studying a metal or a giant interface, stick with PBE+U. Have you had success (or nightmares) running HSE06? Let me know in the comments below. And yes, your SCF will oscillate on the first try—check your mixing parameters. hse06 vasp
In this post, I’ll break down what HSE06 is, how to set it up in VASP, and when it’s actually worth the pain. HSE06 is a screened hybrid functional. It mixes 25% exact (Hartree-Fock) exchange with 75% PBE exchange at short range, while keeping PBE correlation. KPOINTS: Automatic generation 0 Gamma 4 4 2
Enter (Heyd-Scuseria-Ernzerhof). This hybrid functional has become the gold standard for "affordable accuracy" in solid-state physics. But let’s be real—it comes at a computational cost. Let me know in the comments below
SYSTEM = ZnO HSE06 ENCUT = 520 ISMEAR = -5 # Tetrahedron method for DOS SIGMA = 0.05 PREC = Accurate LHFCALC = .TRUE. HFSCREEN = 0.2 AEXX = 0.25 GGA = PE ALGO = Damped TIME = 0.4
If you have spent any time running density functional theory (DFT) calculations, you know the drill: PBE (Perdew-Burke-Ernzerhof) is fast, reliable, and often... wrong. It systematically underestimates band gaps, over-delocalizes electrons, and struggles with strongly correlated materials.
In older VASP versions (pre-6), you needed LHFCALC = .TRUE. and HFSCREEN = 0.2 . In VASP 6+, you can also use HSE06 as a pseudopotential flag, but the manual INCAR approach is safer. Step 2: FFT grids and precision Hybrid functionals are sensitive to the real-space grid. Use high precision: